
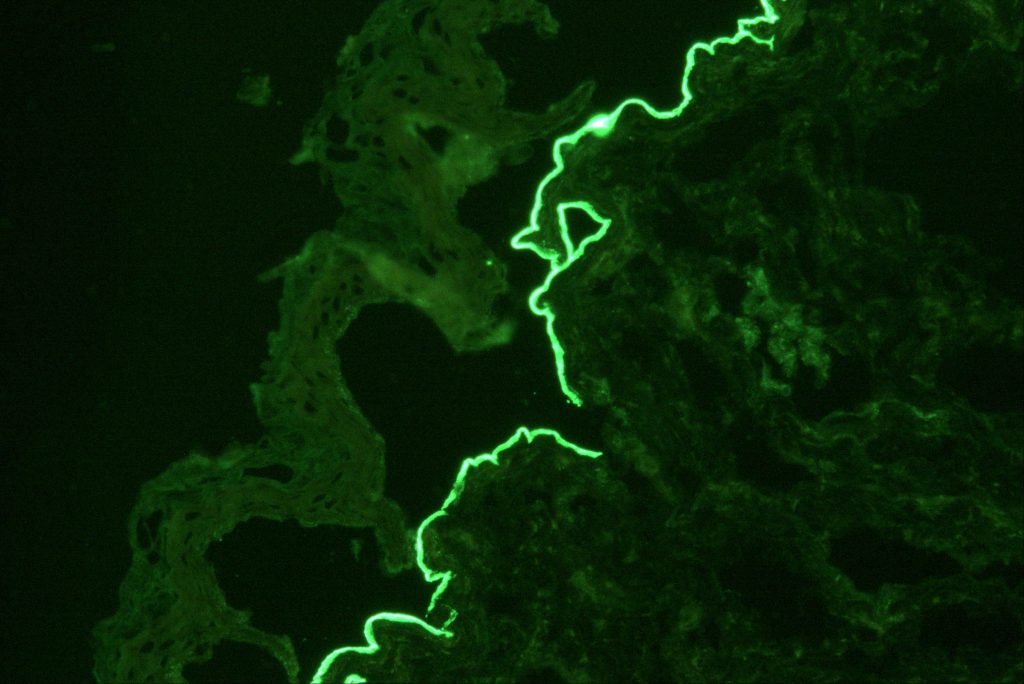
Highly accurate IIF serological assay for Laminin 332 Mucous Membrane Pemphigoid reduces the wait for confirmed diagnosis from months or years to 72 hours or less
On July 27, 2022 KSL Beutner Laboratories (Beutner), a global leader in immunologic testing for the diagnosis of bullous, vascular, connective tissue and inherited skin diseases, announced the launch of a first-to-market indirect immunofluorescence (IIF) serum blood test in the U.S. that positively identifies laminin 332, an antigen associated with the chronic, debilitating autoimmune disease mucous membrane pemphigoid (MMP).
Without a definitive interpretation, patients experience considerable pain and suffering due to misdiagnosis and treatment delays. Long sought after by oral pathologists, oral surgeons, periodontists and dentists, in addition to dermatologists, Beutner’s laminin 332-specific serological assay reduces the time to confirmed diagnosis from as much as two years to 72 hours or less.
“The development and launch of Beutner’s much-anticipated serological assay for laminin 332 MMP builds on the pioneering work of Dr. Ernst Beutner, the father of immunodermatology,” said Dr. Lakshmanan Suresh, Technical Director at Beutner and Chief Medical Officer of KSL Diagnostics, Inc. (www.ksldx.com), which counts Beutner among its clinical laboratories. “The innovative methods he established for diagnosing autoimmune blistering diseases are used worldwide, and we are proud to continue his legacy.”
MMP is an autoimmune blistering disease characterized by multisite lesions on mucous membranes. Anti-laminin 332 MMP lesions often scar and can lead to serious complications depending on the mucosal surfaces affected. Oral mucosa — gums, inner lining of the cheeks and lips, palate and tongue — are involved in 80-90% of cases. Scarring of ocular mucosa, present in half of patients, may lead to blindness. The disease can also impact mucous membranes in the nose, throat, genitals and anus, causing severe, irreversible damage. Another complication in 25-30% of patients is increased risk of cancer malignancies, primarily adenocarcinoma in the gastrointestinal and genital mucosa and lungs. The condition can be fatal if left untreated. Considered a rare disease of unknown cause, MMP occurs mainly in people between ages 60 and 80 and, infrequently, in children. Women are affected twice as often as men.
“The first assay of its kind available in the U.S., Beutner’s IIF serum blood test verifying the presence of specific autoantibodies for laminin 332 MMP now enables clinicians to accurately identify this hard-to-diagnose disease much faster,” said Dr. Raminder Grover, Laboratory Director at Beutner. “This in turn allows for available therapies to be started much earlier to alleviate patients’ significant ongoing discomfort and spare them the long-term medical complications of this devastating disease.”
The most common autoantigens associated with MMP are BP180 and laminin 332. About one-third of patients are afflicted with the laminin 332 variant. Since 2002, blood tests to detect BP180 have been widely available, but a laminin 332 serum test has not been approved in the U.S. until now. Anti-laminin 332 MMP cannot be differentiated from other forms of the disease based on clinical examination. It can only be distinguished by a serological test for IgG antibodies of the variant. Because it mimics other diseases in the mouth, patients may suffer increasing pain and decreased quality of life for six months to two years before obtaining conclusive test results. Reaching a positive diagnosis as soon as possible is critical so that physicians can begin treatment.
Confirming laminin 332 MMP relies on several laboratory tests offered by Beutner: first, a direct immunofluorescence (DIF) microscopy of a skin biopsy to detect tissue-bound immunoreactants. Although DIF is the gold standard for investigating all forms of MMP, it does not always differentiate between variants. Next, indirect immunofluorescence is applied to identify circulating antibodies targeting the autoantigens in the basement membrane zone (BMZ) of the skin. This type of analysis is done in two parts – an IIF test on salt-split skin and an IIF serum test on transfected cells for laminin 332 IgG/IgG4 antibodies using a EUROIMMUN assay. Beutner’s IIF serological assay has a sensitivity of 84% and a specificity of 99%.
Beutner recently received approval from the New York State Department of Health to perform the laminin 332 IIF blood test for U.S. customers at its Buffalo, NY lab.
Dr. Ernst H. Beutner, a co-founder of Beutner Laboratories, pioneered the DIF test 40 years ago after he and his associates at the University at Buffalo (UB) discovered the role of autoimmunity in pemphigus and pemphigoid. Development of defined, quantified immunofluorescent methods now used worldwide for the investigation of autoimmune skin diseases began with studies at UB led by senior researchers from Beutner, UB and Harvard University.
KSL Beutner Laboratories provides anti-laminin 332 IIF serological testing as a service, as well as several other assays for laminin 332 MMP and for the BP180 variant of MMP. Tests can be ordered at https://www.beutnerlabs.com/.





